In the European Union (EU), a Risk Management Plan (RMP) is submitted as part of the dossier for initial marketing authorization of a medicinal product or with an application involving a significant change to an existing marketing authorization. A comprehensive revision of the EU Guideline on Good Pharmacovigilance Practices (GVP) Module V—Risk Management Systems (Revision [Rev] 2), adopted in March 2017, provides a framework for developing more focused, actionable, and risk-proportionate RMPs. This paper describes the Janssen experience with the interpretation and application of GVP Module V (Rev 2) regarding the evaluation of safety concerns in an RMP.
Janssen convened a cross-functional working group to promote consistent interpretation of the GVP Module V (Rev 2) guidance across therapeutic areas. The group created 3 algorithms to support implementation of the guidance related to removal or reclassification of safety concerns by product-specific RMP teams.
Following implementation of the GVP Module V (Rev 2) guidance, the algorithm-driven process led to a substantial decrease in the number of safety concerns for most products. With few exceptions, EU health authorities agreed with the proposed safety concern removals or reclassifications, resulting in RMPs that were focused on only those safety concerns that required further characterization or specific risk minimization.
The algorithm-driven process allows for consistent interpretation and application of the GVP Module V (Rev 2) guidance, which enables product teams to develop an actionable RMP using a thoughtful, evaluative, science-based approach that considers all available evidence.
In the European Union (EU), a Risk Management Plan (RMP) must be submitted as part of the dossier for an initial marketing authorization application, with an application involving a significant change to an existing marketing authorization, or at the request of either the European Medicines Agency (EMA) or competent authority in a Member State [1,2,3,4,5]. The purpose of an RMP is to describe the risk management system for a medicinal product, with a focus on appropriate risk management planning throughout the product life cycle. To this end, an RMP documents the safety profile of a product, emphasizing (1) safety concerns requiring further evaluation and/or risk minimization, (2) pharmacovigilance (PV) activities to characterize the safety concerns, and (3) measures intended to prevent or minimize harm to patients [1,2,3,4,5]. Accordingly, an RMP is composed of 3 core elements: a Safety Specification, a PV Plan, and a Risk Minimization Plan [2, 3].
The RMP Safety Specification describes the safety profile of a product, i.e., what is known about the Important Identified Risks (IIRs), Important Potential Risks (IPRs), and Missing Information (MI) [2,3,4, 6]. An identified risk is defined as an undesirable clinical outcome for which there is adequate scientific evidence of a causal relationship with the medicinal product [3, 4]. A potential risk is defined as an undesirable clinical outcome for which there is scientific evidence to suspect the possibility of a causal relationship with the product, but the evidence is insufficient to confirm a causal relationship. Furthermore, the RMP is expressly focused on those identified and potential risks that are considered important, i.e., those risks that could impact the benefit-risk balance of the product or have implications for public health [3]. Missing Information refers to insufficient knowledge regarding the safety of a product for certain anticipated utilization (e.g., long-term use) or for use in specific patient populations. The IIRs, IPRs, and MI of a product are collectively known as safety concerns [2,3,4, 6].
The RMP PV Plan outlines activities aimed at further characterizing and quantifying the important risks of a product, identifying new important risks, and aggregating knowledge to address MI. In addition, the PV Plan includes specific activities designed to evaluate the effectiveness of additional Risk Minimization Measures (RMMs) captured in the Risk Minimization Plan. The PV Plan consists of 2 types of activities: (1) routine PV activities beyond adverse reaction reporting and signal detection and (2) additional PV activities (Table 1) [2,3,4,5].
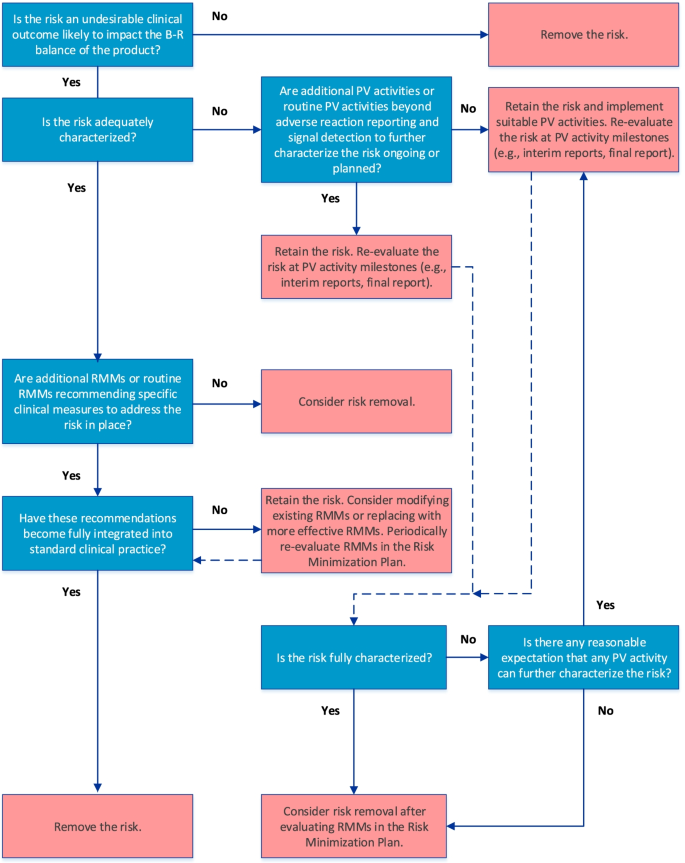
The final algorithm for evaluating IIRs is depicted in Fig. 1. GVP Module V (Rev 2) defines IIRs as undesirable clinical outcomes that are likely to impact the benefit-risk balance of a product and for which there is sufficient evidence that they are caused by the product [3, 4]. In keeping with the overall objective of an actionable RMP, only IIRs that require further characterization (e.g., to determine frequency, severity, seriousness, or outcome of the risk under normal conditions of use or to identify populations particularly at risk) and/or specific RMMs should be included in the RMP.
The first step in the algorithm establishes whether the IIR fulfills the GVP Module V (Rev 2) definition. If it does, the next step in the algorithm, which considers whether the IIR is adequately characterized, is applied. Such characterization may be achieved through routine PV activities beyond adverse reaction reporting and signal detection and/or additional PV activities (Table 1). If the IIR requires further characterization, and if activities to address this are already included in the PV Plan, the IIR should be retained and re-evaluated at key PV activity milestones (e.g., interim or final study reports). Although not specified in the algorithm, if existing PV activities are deemed inadequate at the time of evaluation, they should be modified or replaced. If no activities to investigate the IIR are ongoing or planned despite the need for further characterization, suitable PV activities should be implemented.
After the need for further characterization of the IIR is determined, the next step in the algorithm considers whether routine RMMs recommending specific clinical measures to address the risk and/or additional RMMs (Table 2) are in place. If the IIR is considered fully characterized, it should be retained in the RMP only if RMMs in the Risk Minimization Plan have not yet been fully integrated into standard clinical practice; otherwise, the IIR should be removed from the RMP. The RMMs should be re-evaluated periodically for effectiveness and the need for continued implementation, as suggested in the GVP Module V (Rev 2). If necessary, the RMMs should be modified or replaced with more effective measures.
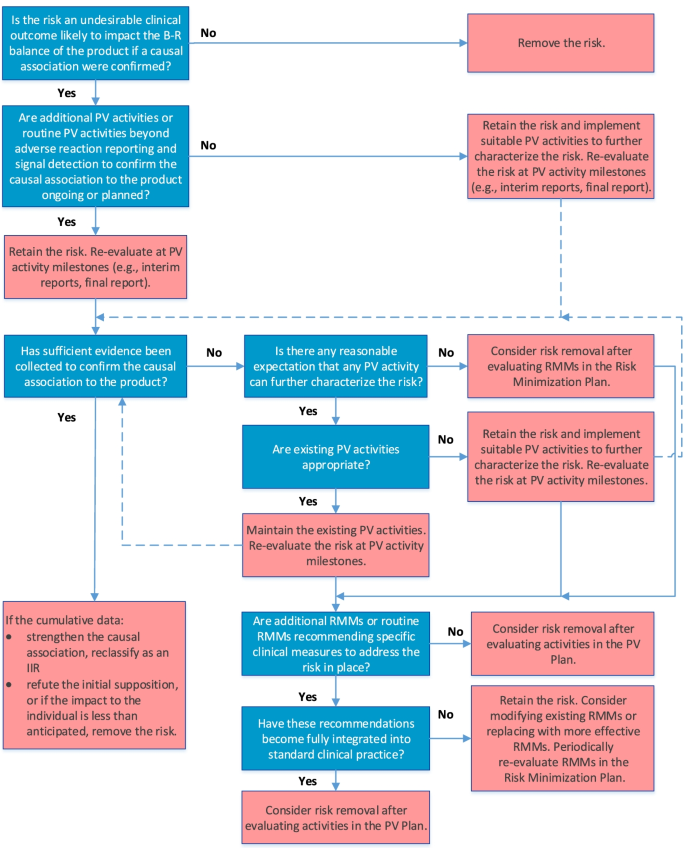
The final algorithm for evaluating IPRs is depicted in Fig. 2. GVP Module V (Rev 2) defines IPRs as undesirable clinical outcomes with a suspected causal association to the product, which, if confirmed, would likely impact the benefit-risk balance of the product [3, 4]. Accordingly, the first question in the IPR algorithm establishes the likelihood of the potential risk to impact the benefit-risk balance of the product if a causal association to the product were confirmed; only those potential risks for which the answer is “yes” are considered IPRs and should be retained in the RMP.
Because IPRs usually require further evaluation as part of the PV Plan, the algorithm next considers whether routine PV activities beyond adverse reaction reporting and signal detection and/or additional PV activities to investigate the IPR are ongoing or planned. If they are not, suitable PV activities to characterize the risk further should be implemented. If PV activities are already included in the PV Plan, the IPR should be re-evaluated at key PV activity milestones to determine whether sufficient evidence has been obtained to confirm or refute causality. If existing PV activities are deemed inadequate at the time of evaluation, they should be modified or replaced. Moreover, if cumulative data confirm a causal relationship, the IPR should be reclassified as an IIR; conversely, if cumulative data disprove a causal relationship, or if the data suggest that the impact to the benefit-risk balance is less than anticipated (i.e., the risk is not “important”), the risk should be removed from the RMP.
If it is determined that no PV activity could further characterize the IPR, the IPR may be removed from the RMP if no measures to minimize the risk are required in the Risk Minimization Plan.
If routine RMMs recommending specific clinical measures to address the risk and/or additional RMMs (Table 2) are in place, they should be evaluated to determine whether they have become integrated into standard clinical practice or require modification or replacement with more effective strategies. As for IIRs, the RMMs should be evaluated periodically for effectiveness and the need for continued implementation.
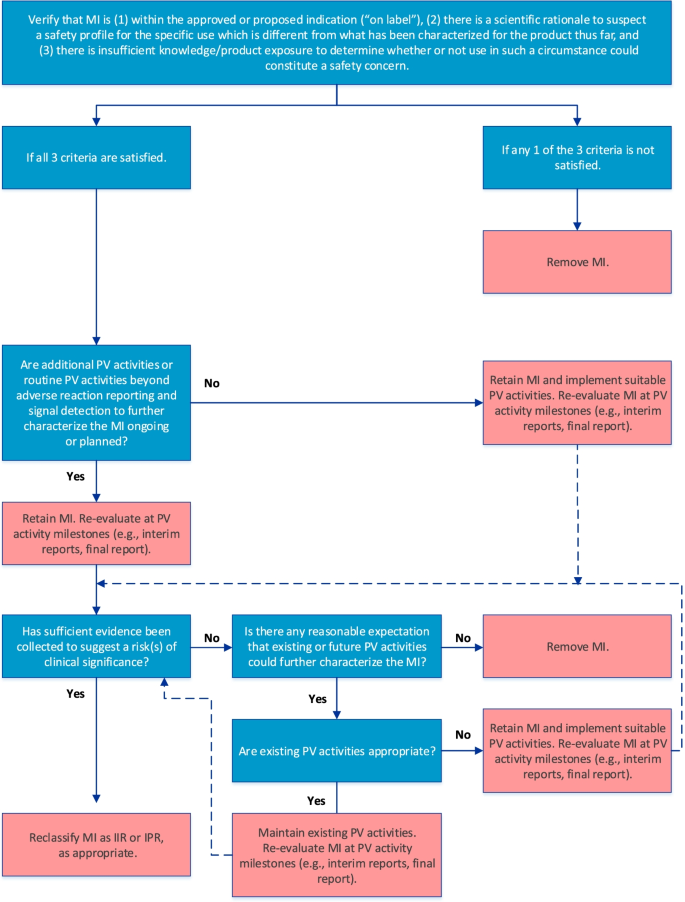
The final algorithm for evaluating MI is depicted in Fig. 3. GVP Module V (Rev 2) defines MI as gaps in knowledge about the safety of a product for certain anticipated utilization or for use in particular patient populations for which there is insufficient knowledge to determine whether the safety profile differs from that characterized so far [3, 4]. For the purposes of risk management planning, MI should be included as a safety concern only if the following 3 criteria are satisfied, as stipulated in the first step of the algorithm: (1) the MI is within the approved or proposed indication as per the product label, (2) there is a scientific rationale to suspect a different safety profile, and (3) there is insufficient product knowledge or exposure to determine whether use of the product in a particular setting or patient population might be associated with risks of clinical significance.
The RMP should include a strategy for obtaining information on the benefit-risk balance where areas of MI exist, which may be achieved through routine PV activities beyond adverse reaction reporting and signal detection and/or additional PV activities. Therefore, the next step in the algorithm confirms whether such activities are already included in the PV Plan. If they are not, suitable PV activities should be implemented; otherwise, the MI should be re-evaluated at key PV activity milestones to determine whether sufficient evidence has been obtained to suggest an important risk, in which case reclassification as an IIR or IPR may be warranted. If the cumulative evidence suggests that the safety profile is not different, the MI should be removed from the RMP. Moreover, if, after a reasonable amount of time on the market, it is determined that no existing or future PV activities could further characterize the safety profile of the product with respect to the MI, the MI should be removed from the RMP.
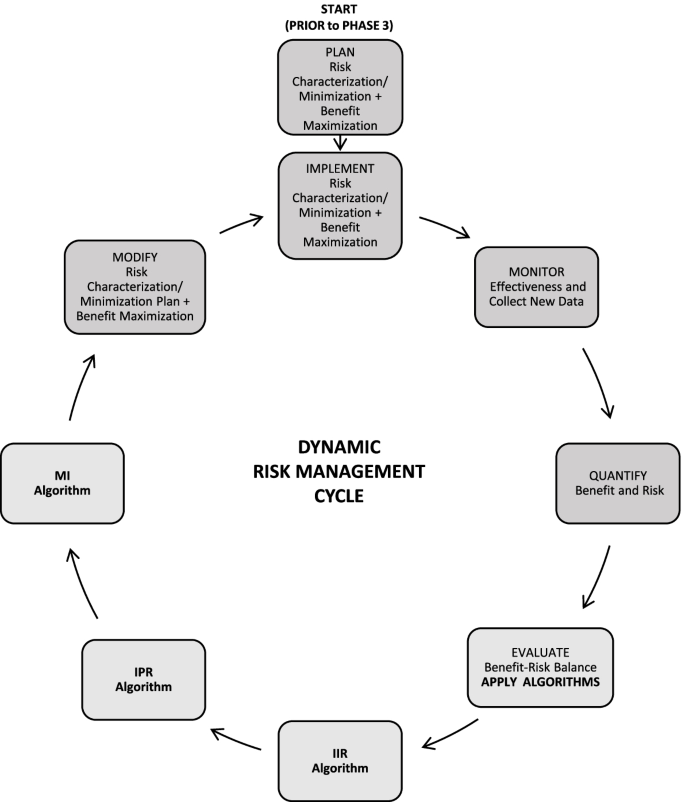
The Janssen working group prepared an informational pack consisting of the final algorithms (Figs. 1, 2, and 3) with operational footnotes (Tables 1 and 2), along with a training slide deck and high-level coaching tools (Figs. 4 and 5) to provide additional guidance to product-specific RMP teams [3, 4]. Figure 4 depicts the dynamic nature of the RMP process and includes specific points in the product life cycle at which the Safety Specification should be re-assessed and where the Janssen algorithms fit within the process. Figure 5 lists the spectrum of sources (regulatory history, clinical or observational trial data, product information, trending analyses from the global safety database, postmarketing safety data, results from additional PV activities, and the scientific literature) that product-specific RMP teams should consider when applying the algorithms.
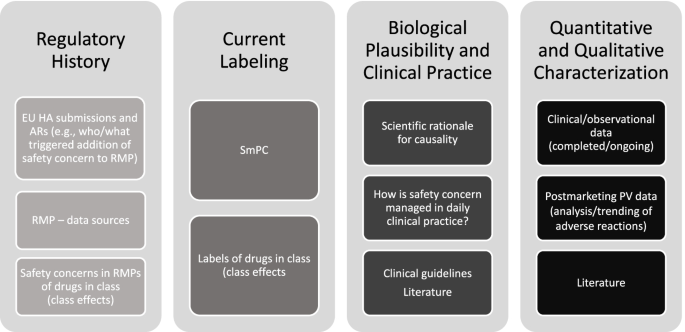
Prior to re-evaluating RMPs, product-specific RMP teams are advised to review relevant EU health authority (HA) assessment reports and requests related not only to the product RMP under evaluation, but also to RMPs for other Janssen products and non-Janssen products in the same class. For each safety concern proposed for removal or reclassification utilizing the final algorithms, product-specific RMP teams are required to provide adequate justification along with appropriate reference to the safety data, EU HA assessment report, and EU HA request (including reference to the relevant procedure).
To assess the performance of the algorithm-driven process for RMP re-evaluation, information from RMP Annex 8 (Summary of Changes to the Risk Management Plan Over Time) was analyzed to determine the number of safety concerns in each product-specific RMP before application of the algorithms and the number of safety concerns removed or reclassified following EU HA assessment and approval. If more granular information was needed, additional details were obtained from the respective product-specific RMP team.
Between March 2018 and March 2020, a total of 26 RMPs pertaining to 22 Janssen products were re-evaluated via the algorithm-driven process and subsequently submitted to EU HAs for assessment. For 4 out of 22 of these products, RMPs were re-evaluated in 2 separate procedures after new data became available. Results for the different RMPs for these 4 products were integrated per product for the purpose of this analysis (i.e., a total of 22 RMPs were included in the final analysis). Prior to application of the algorithms, the 22 RMPs evaluated included a median of 18.5 (interquartile range [IQR] 15.0, 24.0) safety concerns. After application of the algorithms and following final assessment by EU HAs, the median number of safety concerns per RMP dropped to 3.5 (IQR 1.0, 9.0). As shown in Fig. 6A, the median % reduction in safety concerns was 82.4% (IQR 55.0%, 93.8%). Median % reductions were similar across all 3 types of safety concerns (IIR: 84.9% [IQR 25.0%, 100%]; IPR: 86.9% [IQR 42.9%, 100%]; MI: 75.0% [IQR 60.0%, 100%]).
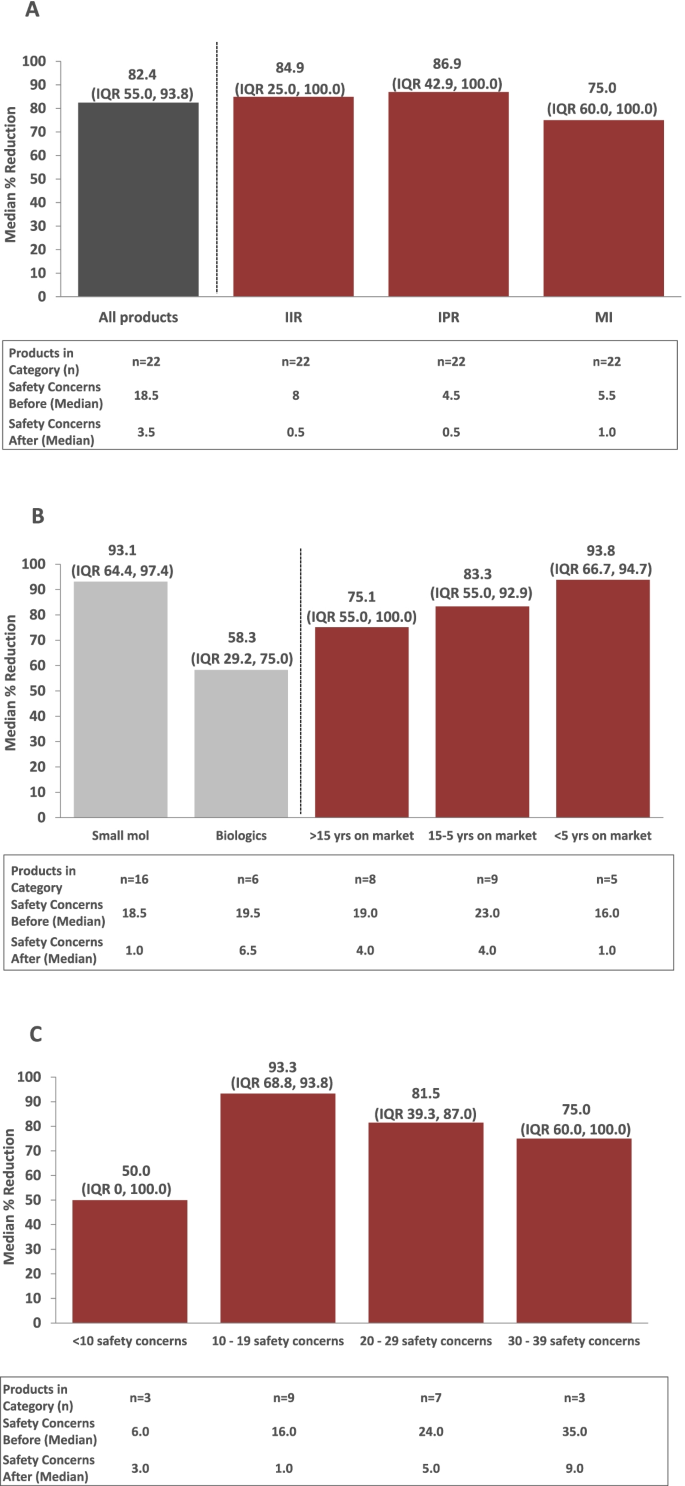
Eight of the 22 RMPs evaluated involved products that were marketed for more than 15 years (Fig. 6B). Nine RMPs involved products that that were on the market between 5 and 15 years. The remainder of RMPs (n = 5) involved products that were launched within the preceding 5 years. Median % reductions in safety concerns were generally similar across RMPs, irrespective of length of time since the product received marketing approval (75.1% to 93.8%; Fig. 6B). When the median % reduction in safety concerns among RMPs was analyzed by product type (small molecule versus biologic; n = 16 vs. n = 6, respectively; Fig. 6B), reductions tended to be greater among RMPs for small molecules (93.1% [IQR 64.4%, 97.4%]) compared with biologics (58.3% [IQR 29.2%, 75.0%]); this was presumably driven by 2 RMPs for biologics that included only a few safety concerns.
An analysis of the median % reduction in safety concerns by the number of safety concerns included in the RMP prior to application of the algorithms supported this conclusion. RMPs with 10 to 19, 20 to 29, and 30 to 39 safety concerns prior to application of the algorithms had greater median % reductions in safety concerns (93.3% [IQR 68.8%, 93.8%]; 81.5% [IQR 39.3, 87.0%]; and 75% [IQR 60.0%, 100%], respectively) compared with those with < 10 safety concerns (50%; [IQR 0, 100]), indicating that the number of safety concerns at the outset of the exercise may have played a role (Fig. 6C). However, the number of products with < 10 safety concerns was small (n = 3).
The reasons driving the decision for removal or reclassification varied by type of safety concern, as shown in Table 4. For important risks (IIRs and IPRs), common reasons for removal were that the risk was fully characterized, with no additional PV activities ongoing or planned, or that further characterization was not anticipated, e.g., due to the rarity of the event. Other frequent reasons for removing important risks were that no additional RMMs were in place or that, if they were in place, the additional RMMs were no longer required because guidance for management of the risk had been fully integrated into clinical practice. A common reason for the removal of safety concerns classified as MI was that they no longer met the revised GVP Module V (Rev 2) definition of MI; for example, the MI was not within the approved indication or there was no scientific rationale to suspect a different safety profile.
Table 4 Rationales for successful reclassification or removal of a safety concernOverall, EU HAs were in agreement with the removals or reclassifications of safety concerns proposed by Janssen. In some cases, EU HAs suggested modifications beyond those proposed or specifically described in GVP Module V (Rev.2). For instance, it was requested that safety concerns with a similar etiology that could be addressed through the same measures (e.g., subtypes of malignancies or infections) be grouped together as a single safety concern. In other cases, in particular for established products, EU HAs requested harmonization of safety concerns across an entire therapeutic class.
Marketing authorization applicants, marketing authorization holders, and EU HAs have a shared interest in minimizing risks for any given medicinal product and improving the benefit-risk balance for patients within the context of risk management planning. Ideally, risk management planning should be targeted and based upon a risk-proportionate set of activities that directs resources to areas where the need for additional information and risk minimization is greatest, without placing undue burden on healthcare providers and patients. The implementation of GVP Module V (Rev 2) in 2017 provided the framework to achieve this goal. Only those safety concerns that are likely to impact the benefit-risk balance and that require active management in terms of further characterization and/or specific risk minimization should be included in the Safety Specification of the RMP.
GVP Module V (Rev 2) stands out from previous RMP guidance, which promoted lengthy lists of safety concerns that tended to increase in number over time. The RMP is no longer regarded as a “safety haven” characterized by an all-inclusive, but not actionable, list of safety concerns. Instead, the RMP is a living document that is expected to be re-evaluated and fine-tuned over the product life cycle in light of increasing product knowledge. Importantly, inclusion of any safety concern in the RMP should be a thoughtful and data-driven process.
While GVP Module V (Rev 2) represents a welcome paradigm shift in risk management planning over the product life cycle, it also poses a challenge to marketing authorization applicants, marketing authorization holders, and EU HAs alike, all of whom are tasked with applying the guidance in a rational and consistent manner.
In contrast to its predecessor, GVP Module V (Rev 2) is less didactic and allows for greater interpretation, with consistent implementation of the guidance largely left to the discretion of marketing authorization applicants and marketing authorization holders. In response to this challenge, Janssen developed a new approach, which includes the algorithms described herein, that may be applied reliably across product portfolios.
The algorithm-driven process developed by Janssen for the removal and reclassification of safety concerns is a structured, data-driven, and regulatory-compliant approach that promotes consistent interpretation and application of GVP Module V (Rev 2) across products regardless of product type and life-cycle stage. Each RMP is periodically re-evaluated as product knowledge is accrued and/or relevant PV activity milestones are achieved.
Upon incorporation of the EU HA assessment, application of the algorithm-driven process to 22 Janssen product RMPs resulted in a median % reduction of 82.4% (IQR 55.0%, 93.8%) in the total number of safety concerns. In the majority of cases, EU HAs accepted the proposals for the removal or reclassification of safety concerns.
Although the algorithms provide the benefit of a consistent, science-driven, and regulatory-compliant approach, they cannot address those limitations that apply to risk assessment in general. Supportive data on adverse reactions are collected from multiple sources, with varying levels of quantity and quality, complicating the evaluation of their seriousness, relatedness, and relevance. Not every adverse reaction is considered an important risk for the product in a given therapeutic context. Thus, careful deliberation and documentation of decisions are critical.
Prior to applying the algorithms to an RMP, vigorous data collection (as outlined in Fig. 5) is recommended so that re-evaluation of the RMP can be accomplished as efficiently as possible and any proposed revisions may be appropriately justified. As a general rule, safety concerns with no associated activities in the PV and Risk Minimization Plans and for which there is no reasonable expectation that further investigation can provide additional characterization should be considered for removal from the RMP.
For mature products that are part of a therapeutic class with an established safety profile, EU HAs may request harmonization of safety concerns across the class. Also, risks that share the same etiology and that can be addressed through the same PV activities and/or RMMs may be grouped under a single safety concern.
In the past, marketing authorization applicants and marketing authorization holders tended to include populations excluded from the clinical development program as MI in the RMP, based solely on an absence of data. This practice contrasts with the requirements for MI as set forth in GVP Module V (Rev 2), i.e., if postauthorization use in the unstudied population is expected, the population must be included within the target indication and a different safety profile in the population must be suspected. A scientific rationale is needed for any population included as MI in the RMP (see Table 4 for examples).
Consequently, use in an unstudied population no longer automatically constitutes a safety concern. For example, use in patients with hepatic or renal impairment should be included as MI only if the product is metabolized hepatically or excreted renally and, consequently, different risks could be anticipated in these patients. Similarly, use in pregnant women should be included as MI only if women of childbearing potential are within the approved indication. Off-label product use, including use in patients for whom the product is contraindicated, is no longer considered MI. Rather, if the product is likely to be used outside the approved indication and an important risk arising from such use is anticipated, the risk should be included as a safety concern only if it is not already an IIR or IPR for the product. It should be made clear that this safety concern is associated specifically with off-label use. Any MI included in the RMP should be reassessed at PV activity milestones, as this designation may no longer apply as postmarketing experience increases and additional data become available.
In addition to evaluating safety concerns for possible removal or reclassification, it is important that product-specific RMP teams assess the relative burden versus benefit of the associated risk management activities. Understanding and assessing the burden on patients, healthcare professionals, and the wider healthcare system is an important consideration in RMP preparation and revision [7].
As GVP Module V (Rev 2) represents a markedly different approach to risk management planning compared with earlier guidance, continued learning on the part of marketing authorization applicants, marketing authorization holders, and EU HAs is to be expected. Engaging in an open dialogue, addressing potential inconsistencies, and questioning ambiguous decisions when appropriate has proven helpful in this learning process.
The algorithm-driven process allows for consistent interpretation and application of the GVP Module V (Rev 2) guidance, which enables product teams to develop an actionable RMP using a thoughtful, evaluative, science-based approach that considers all available evidence. It is important that experience with GVP Module V (Rev 2) be shared among Industry stakeholders and EU HAs to move the field of risk management forward. By publishing the Janssen experience, other stakeholders may glean insights to support the development or refinement of their own RMP processes. Sharing lessons learned will ultimately strengthen the approach to risk management, to the benefit of the patients.
All data analyzed and generated during this study are included in this publication.
Cathelijne de Gram, EMEA Regulatory Affairs, Janssen Biologics BV, The Netherlands; Carla Morris, Janssen Biologics BV, The Netherlands; Mark Caldwell, Janssen-Cilag Ltd., United Kingdom; Michael Clark, Johnnie Lee, Angela Jenkins-Zoller, and Alianu Akawung, Janssen Research & Development, LLC, United States contributed to the original flow chart designs, piloted the RMP processes, assisted with the quantitative performance analyses, and/or critically reviewed the manuscript content. Marie-Laure Ruhf, Kenneth E. Robertson, and Holly Capasso-Harris, PhD of Synchrogenix, LLC, a Certara company, provided medical writing assistance that was funded by Janssen Biologics BV.
This work was supported by Janssen Biologics BV.